Abstract
Parkinson’s disease (PD) is a neurodegenerative disorder marked by the loss of dopaminergic neurons in the substantia nigra. Despite progress, the pathogenesis remains unclear. Human midbrain organoids (hMLOs) have emerged as a promising model for studying PD, drug screening, and potential treatments. This review discusses the development of hMLOs, their application in PD research, and current challenges in organoid construction, highlighting possible optimization strategies.
Introduction
Parkinson’s disease (PD) is one of the most common neurodegenerative disorders associated with movement disabilities, affecting more than 6.1 million people worldwide1,2, with the mean age of onset at 553. Clinically, as a debilitating neurological disorder, PD patients mainly present with motor symptoms such as resting tremor, bradykinesia, rigidity, postural instability, loss of coordination and shuffling or freezing gait4,5,6. Non-motor symptoms may also present including depression, anxiety, constipation, sleep disturbances, hyposmia, paresthesia, and cognitive abnormalities7,8,9,10. The key histopathological hallmark of PD is the gradual loss of midbrain dopaminergic (mDA) neurons and the presence of intraneuronal protein inclusions named “Lewy Bodies” (LB) in substantia nigra (SN)3,10,11,12. Composed of abnormal α-synuclein (α-syn) protein aggregations13, LBs-associated pathology has been attributed to mitochondrial metabolism alteration and proteasomal and autophagy-lysosomal dysregulation, which ultimately bring about the death of mDA14,15.
Up to now, there are three prevalent tools to explore the underlying mechanism of PD, including the post-mortem brain tissue from PD patients, animal models and in vitro cell models. The post-mortem brain of PD patient is an ideal source for the PD analysis which directly reflect the actual inner environment. The use of human brain tissue, however, is strictly restricted by practical constraints16,17,18,19,20,21. Furthermore, post-mortem brain tissue may have undergone irreversible changes during the process of death that limit its utility for the study of PD16. Animal models of PD can be further divided into two groups, the toxin-based model and gene-based model3. By introducing neurotoxins such as 6-hydroxydopamine (6-OHDA), 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), paraquat, and rotenone, toxin-based models suffer from overloaded oxidative stress which provokes the speedy degeneration of mDA that mimic the sporadic PD17,18. On the other hand, gene-based models are built via overexpress or knockdown/knockout certain genes such as LRRK2, Parkin, DJ-1, and PINK1, to study the molecular mechanism of their involvement in PD pathology18,19,20. However, the value of animal models is limited because of their inherent differences from humans21,22. In vitro cell models are one of the most easily obtained approaches to investigate PD pathogenesis, with the advantage of shorter time consumption and smaller inter-batch differences, not until the advent of iPSCs technology revolutionized the modeling of human diseases including PD23. Combined with genome editing techniques24, this approach offered the opportunity to give rise to disease-relevant neuronal subtypes that retain the certain genetic background of the patient. Nonetheless, current approaches for mDA neuron generation are typically based on two-dimensional (2D) conditions, however, mDA neurons yielded from iPSCs without other cell co-cultivation clearly neglect the fact that in vivo neuron growth and maturation does not work on their own25,26,27. What’s more, the granular pigments named neuromelanin (NM) synthesized by DA in human SN has not been seen in the 2D-derived mDA neurons28,29,30,31.
Thus, an appropriate in vitro model that reflects human neurobiology to advance our insight of the pathology of PD is urgently needed. The introduction of midbrain organoids allowing us to overcome some of the obstacles, given either by the use of animal models, such as species specificity of cellular pathways, ethical issues and the limited availability of post-mortem human brain tissue. This review briefly summarizes the development of midbrain organoids since their inception and focuses on their PD-related application, providing new hints for further condition optimization and clinical transformation of midbrain organoids.
The midbrain organoids and Parkinson’s disease
Over the last decade, three-dimensional (3D) organoid technology has become popular in stem cell research. Organoids are miniature in vitro 3D cellular clusters generated from iPSCs or isolated organ progenitors32,33,34,35,36. Due to their capacity for self-renewal and self-organization, organoids can mimic many aspects of in vivo organs. Organoids have been generated that model various parts of the brain, including the forebrain, midbrain, cerebellum, cortex, and hippocampus37,38,39. Various methods are used to generate these organoids, many of which aim to model the development of the human brain. In addition, brain organoids have been used to recapitulate human disease. In this context, the rise of human stem-cell derived brain 3D organoid cultures, which recapitulate features of the brain’s composition, organization, and function40, has led to significant advances in our understanding of neurodevelopment and in disease modeling. Although midbrain and mDA markers have been found to spontaneously arise in non-directed whole brain organoids41, the proportions of cells expressing such markers tend to be small and highly variable, thus warranting the development of more directed differentiation protocols. Some approaches have led to the development of “neurospheres,” which contain an increased proportion of DA neurons, along with excitatory, inhibitory neurons as well as glial cells42. Most efforts have been directed at specifically reproducing mesencephalic development in the dish, in order to generate mDA neurons in representative human “midbrain organoid” (hMLO) structures. In general, the development of hMLOs involves two stages, namely floor plate induction and organoid development. By incorporating specific components, iPSCs are differentiated from a 2D state into 3D spheres with midbrain-specific markers (Fig. 1).

Schematic diagrams illustrating the overall strategy to generate hMLOs. Differential interface contrast (DIC) images illustrate the typical morphology of cells at each stage. SBNC: SB431542, Noggin, and CHIR99021; SF: SHH-C25II and FGF8; BGAC: BDNF, GDNF, ascorbic acid, and db-cAMP. This image was independently created by us using Adobe Illustrator. All elements are original, and no third-party materials were used.
Tieng et al.43 were the first to adapt a widely-used 2D differentiation protocol25 to 3D suspension through the use of microwells to create homogeneously sized embryonic bodies, which were then placed on an orbital shaker for 3 weeks, before being seeded and grown at air-liquid interface. Although the suspension-culture phase of their protocol was short, they proved that such an approach could efficiently generate mDA progenitor cells (∼80% of all cells expressed FOXA2 and LMX1A) as well as TH-expressing cells after only 3 weeks. Following these results, 3 new protocols were published within 1 year39,44,45, describing the generation and long-term maintenance of hMLO (up to 5 months). These papers were the first to provide in depth characterization of the model, and proof that these organoids could be maintained in long term cultures in order to favor neuronal maturation. Although each protocol presents differences in timing, specific molecules used and their concentrations, these approaches mainly rely either on the sequential44 or simultaneous39,43,45 use of morphogens to induce midbrain floor plate identity, as described earlier. These organoids developed features of organization similar to the midbrain floor plate, namely a ventricular zone containing OTX2+FOXA2+ cells, as well as intermediate (LMX1A+, NURR1+) and mantle layers containing progressively maturing neurons (MAP2+, TH+). Several markers of pan-mDA neuronal identity have been consistently observed in hMLO, including the dopamine transporter (SLC6A3/DAT), DOPA decarboxylase enzyme DDC, and TF PITX344,46. Studies also found that NM granules can spontaneously appear in long term cultures, their structures resembling those found in adult human SN tissue. NM-containing cells were found to be enriched in transcripts expressed in A9 SN mDA neurons44,47,48,49. Exogenous DA treatment could also significantly increase the accumulation of NM, suggesting that these granules may indeed be by-products of DA metabolism44. There have been reports demonstrating that transplantation of hMLOs into mouse brains can establish classical substantia nigra-striatum network connections, further confirming the presence of functionally relevant SN components in organoids50. The hMLOs were also found to produce DA, and mDA neurons showed characteristic electrophysiological pacemaker activity which was responsive to the use of D2/D3 agonist quinpirole. Beyond mDA neurons and their progenitors, excitatory and inhibitory neurons43,44 were found in these hMLO, as well as astrocytes and myelinating oligodendrocytes, consistent with the composition of the midbrain44,45.
The construction of hMLOs offers an attractive alternative to 2D culture models, they recapitulate some of the complex characteristics and physiology of the human midbrain that are lacking in 2D culturation. They may also provide the missing link between in vitro 2D models and in vivo animal models. Moreover, the ability to maintain organoids for long-term culture, up to 1 year, means that they may be used for aging studies, and/or long-term drug interventions. Due to the multifactorial nature of neurodegenerative diseases such as PD, the use of single cell type models might limit target discovery and the potential for phenotypic screening. The usage of animal models can also be limited due to species differences51. hMLOs provide a human-based 3D culture system that may combat the limitations of other models in the drug screening process, either. In addition, since iPSC carry the same genetic signature as the patient they are derived from, the generated organoids will express any pathogenic mutations present.
Since the advent of hMLO in 201440, some of the major breakthroughs in the PD field are the results of hMLO usage. Human brain-based models of PD have contributed greatly to our understanding of disease mechanisms and pathogenesis, while also facilitating a means to test viable drug candidates52,53,54. PD treatment is also a promising application for hMLO55.
Application of hMLOs in PD
In the following sections, we will delve into three key aspects of recent advancements in the application of human midbrain-like organoids: constructing PD models; predicting PD toxicity; and developing treatments for PD (Fig. 2).

Three main directions of hMLOs in PD research: PD model construction, PD toxicity predictions and PD treatment. PD model construction involves deriving iPSCs from PD patients or using gene-edited iPSCs to generate midbrain organoids, which are then used to investigate PD-related pathological changes, protein dysfunction, and neuronal degeneration. PD toxicity predictions explore the effects of oxidative stress-inducing compounds and selective toxins that target dopaminergic neurons. PD treatment research includes the extraction of neural stem cells (NSCs) from midbrain organoids for transplantation in mouse models and the selection of drugs that may provide therapeutic benefits. This image was independently created by us using Adobe Illustrator. All elements are original, and no third-party materials were used.
PD model construction
For the past few years, organoids have widely been used in neurodegenerative disease, including Alzheimer’s disease56,57, amyotrophic lateral sclerosis58,59, spinal muscular atrophy60 and Huntington’s disease59. As mentioned earlier, the conventional models for PD studying (in vitro 2D cell models, animal models and the post-mortem brain tissue) all have their unneglected drawbacks, the advent of organoids is considered a major breakthrough in stem cell research and has enabled advancements in the applications of human iPSC for disease modeling. Since then, various methods have been developed to generate models aiming to mimic human midbrain contents.
With the prevalence of hMLOs, they are increasingly used in PD modeling. Similar to the construction of traditional PD animal models, there are 2 popular strategies in PD-related hMLOs induction, which are genetic-based and toxin-based, respectively. The development of hMLOs provides a more advanced model for the in-depth study of PD.
Construct hMLOs carrying monogenic PD disorders
With the advancement of genetic techniques and population studies, over 20 monogenic forms of PD have been described and over 100 loci have been identified as risk factors for PD61,62,63,64, however, most of their pathogenesis are poorly understood. Recent advances in 3D organoid technology offer promise in advancing the understanding of PD on a platform more physiologically44,65,66. The combination of specific genetic mutation with hMLO construction shed light on the further exploration on PD pathogenesis. We have summarized the recent research using hMLOs in the study of PD genetic mutations and their pathogenetic mechanisms in Table 1.
SNCA
SNCA, the first gene associated with familial Parkinson’s disease, encodes the protein α-syn67. Missense mutations in the SNCA gene cause a rare, autosomal dominant inherited form of Parkinson’s disease68,69,70,71,72. Moreover, copy number variations (CNVs) in the SNCA gene were also identified in patients with Parkinson’s disease. Indeed, the clinical phenotype of SNCA duplications resembles typical late-onset sporadic Parkinson’s disease whereas SNCA triplications lead to a more widespread neurodegeneration with early-onset parkinsonism and dementia72,73,74,75,76,77. This implies that there is a direct relationship between SNCA gene dosage and disease severity73,78. Beyond these rare genetically determined forms, the abnormal accumulation of α-syn in the brain is a classical pathological hallmark for a group of related neurodegenerative disorders, collectively referred to as synucleinopathies. To date, the majority of studies examining α-syn pathophysiology have relied on 2D cell culture systems and rodent models78,79. With the advent of human induced pluripotent stem cells (iPSCs) and 3D brain organoids, it is now possible to use patient-derived models to more faithfully reconstitute human brain-region specific features of the disease in vitro.
Up to now, there are four comprehensive studies about building SNCA mutant hMLOs using different cell resourses80,81,82,83. Mohamed et al.81 use three-dimensional midbrain organoids differentiated from iPSCs derived from patients carrying a triplication of the SNCA gene and from CRISPR/Cas9 corrected isogenic control iPSCs. These human midbrain organoids recapitulate key features of α-synuclein pathology observed in the brains of patients with synucleinopathies. The SNCA triplication human midbrain organoids express elevated levels of α-synuclein and exhibit an age-dependent increase in α-synuclein aggregation, hinting that hMLOs carrying SNCA gene multiplication can reliably model key pathological features of PD and provide a powerful system to study the pathogenesis of synucleinopathies. Jo et al.80 further generated and characterized hMLOs from GBA1−/− and SNCA overexpressing isogenic embryonic stem cells by gene-editing technic to investigate genotype-to-phenotype relationships in PD, with the particular aim of recapitulating α-syn-and Lewy body-related pathologies and the process of neurodegeneration in the hMLO model. They identified for the first time that the loss of glucocerebrosidase, coupled with wild-type α-syn overexpression, results in a substantial accumulation of detergent-resistant, β-sheet-rich α-syn aggregates and Lewy body-like inclusions in hMLOs. Becerra-Calixto et al.82 showed that fPD-hMLOs from patients with SNCA triplication spontaneously accumulated pathological α-syn and exhibited significant neurodegeneration. Similarly, Muwanigwa et al.83 found that 3xSNCA hMLOs from PD patients displayed elevated levels of α-syn, progressive loss of dopaminergic neurons, and a senescent-like phenotype in astrocytes.
LRRK2
Missense mutations in the leucine-rich repeat kinase 2 (LRRK2) gene locus are the most common known causes of late-onset familial and sporadic PD84,85. Previous studies have suggested that the LRRK2 G2019S gene mutation is associated with α-synuclein accumulation, mitochondrial dysfunction, and impaired dopamine signaling in the human brain, eventually resulting in the progressive loss of dopamine neurons86,87,88,89. However, a particularly difficult challenge in understanding the role of LRRK2 in PD research has been the generation of models that accurately recapitulate the LRRK2 mutant-associated disease state. For example, animals that harbor genetic mutations mimicking the familial forms of parkinsonism, including LRRK2 mutations, fail to show clear evidence of progressive midbrain dopamine neuron loss or Lewy body formation90,91,93. Another approach that has been taken to model PD is the use of patient-derived iPSCs directed to differentiate into dopamine neurons. These models also show variable dopamine neuron toxicity, but other features of PD pathology, such as Lewy body aggregates, are not as prominent as in the human brain94, and such culture systems are generally immature95.
The first two in-depth reports of PD modeling in hMLO focused on the effects of the LRRK2 G2019S mutation, which has been associated with both sporadic and familial forms of the disease due to its variable penetrance96, and which constitutes the most common genetic risk factor for PD. To do so, the researchers relied on Crispr-Cas9 gene editing to either introduce the mutation in a control iPSC line31, or to combine this with a correction in a mutant patient line97. Smits et al.97 found that while the number of mDA progenitors (FOXA2+TH− cells) was significantly increased after 1 month of differentiation in LRRK2 vs. control hMLO, an apparent impairment of differentiation led to a reduction in the number and complexity of mDA neurons (FOXA2+TH+) after longer periods of culture (day 70). Interestingly, the increase in the number of progenitors was significantly higher in LRRK2 PD hMLO compared to those from controls with the knock-in mutation. This result thus highlights the importance of the genetic background in the penetrance of the LRRK2 G2019S variant96. In line with these findings, Kim et al.31 observed that while LRRK2 G2019S hMLOs were no different in size compared to controls, mDA neurite length and expression of mDA identity markers were decreased (such as TH, DAT, NURR1, PITX3, EN1) by day 60. The LRKK2 hMLO also contained higher levels of phosphorylated α-syn in endosomal compartments, and higher expression levels of markers of mitophagy and autophagy. The authors also identified TXNIP98 as an important mediator of LRRK2-G2019S pathological mechanisms, and proved that knocking-down its expression reversed the accumulation of phosphorylated α-syn. Additionally, a study by Zagare et al.99 explored the LRRK2 p.Gly2019Ser mutation within midbrain organoids, revealing through single-cell RNA sequencing that this mutation induces early neurodevelopmental alterations. Specifically, it affects differentiation patterns and reduces cellular variability, suggesting critical early-stage disruptions in Parkinson’s disease progression.
PINK1
Autosomal recessively inherited mutations in the PINK1 gene typically cause early onset PD100,101. PINK1 has been implicated in the regulation of mitophagy, mitochondrial function and oxidative stress102,103,104,105,106,107,108, however, the underlying mechanisms of PINK1-mediated PD are not fully understood. Pink1 knockout mice do not display reductions in DA neurons in the substantia nigra10. In contrast, PINK1 deficiency in zebrafish results in both reduced numbers of DA neurons in larval and adult zebrafish as well as impaired mitochondrial function and morphology107.
Recent studies employing hMLOs have shed light on the functional roles of the PINK1 gene in Parkinson’s disease. In the research conducted by Eldeeb et al.109, iPSC-derived DA neurons and hMLOs were utilized to investigate the endogenous high-molecular-weight (HMW) PINK1 complex in physiologically relevant models. Their findings revealed that treatment with CCCP and ammonium chloride induced a PINK1- and Tom40-positive 720-kDa HMW complex in wild-type DA neurons and hMLOs, but not in PINK1 knockout models, underscoring the essential role of PINK1 in the assembly of this complex, which is crucial for mitochondrial functionality. Another study by Brown et al.110 explored the impact of PINK1 deficiency on dopaminergic neurogenesis using isogenic midbrain-specific organoids derived from small molecule neural progenitor cells. This study found that PINK1-deficient organoids exhibited a significantly reduced growth rate and impaired differentiation of DA neurons, highlighting the gene’s importance in neurodevelopmental processes related to PD. Together, these studies emphasize the utility of hMLOs in modeling PD at a cellular level, particularly in understanding how genetic variations such as those in PINK1 affect disease progression and neuronal functionality.
GBA1
Mutations in the GBA1 gene are widely recognized as one of the most significant genetic risk factors for PD, particularly associated with the early onset and more severe progression of the disease111,112. The GBA1 gene encodes the lysosomal enzyme GCase, which is crucial for the degradation of glycolipids. Deficiencies or dysfunctions in GCase due to GBA1 mutations result in the accumulation of glycolipid substrates, contributing to lysosomal dysfunction and α-synuclein aggregation, which are hallmark features of PD pathology113,114.
Research utilizing iPSC-derived midbrain organoids has significantly advanced understanding of GBA1 mutations in Parkinson’s disease. Baden et al.115 developed organoids to study mitochondrial dysfunctions associated with GBA1 mutations, demonstrating reduced GCase activity and increased insoluble α-synuclein, which impacts dopamine neuron health. Meanwhile, Rosety et al.116 focused on the neurodevelopmental effects of the N370S GBA mutation, revealing impaired neuronal differentiation and increased oxidative stress, alongside metabolic alterations predisposing cells to neurodegeneration. Collectively, these studies underscore the critical insights provided by midbrain organoids into the complex mechanisms of GBA1 mutations within disease-relevant models.
Other related genes
DNAJC6 encodes auxilin, which acts as a co-chaperone to recruit HSC70 to clathrin-coated vesicles for disassembly117. Homozygous loss-of-function mutations of DNAJC6 have been identified in familial juvenile parkinsonism118,119,120. However, the pathogenic mechanism for PD caused by DNAJC6 mutations remains unclear. Wulansari et al.121 built hMLOs carrying DNAJC6 mutation with key PD pathologic features, i.e., mDA neuron degeneration, pathologic α-synuclein aggregation, increase of intrinsic neuronal firing frequency, and mitochondrial and lysosomal dysfunctions. In addition, neurodevelopmental defects were also manifested in hMLOs carrying the mutations. Transcriptomic analyses followed by experimental validation revealed that defects in DNAJC6-mediated endocytosis impair the WNT-LMX1A signal during the mDA neuron development. Furthermore, reduced LMX1A expression during development caused the generation of vulnerable mDA neurons with the pathologic manifestations. These results suggest that the human model of DNAJC6-PD recapitulates disease phenotypes and reveals mechanisms underlying disease pathology, providing a platform for assessing therapeutic interventions.
Mutations in the E3 ubiquitin ligase (PARKIN), the protein deglycase (DJ-1) and the presumptive cation-transporting ATPase 13A2 (ATP13A2) are inherited in an autosomal recessive fashion and cause completely penetrant early-onset PD in homozygous or compound heterozygous carriers122. It is unclear how dysregulation of these genes results in the relatively selective death of nigral dopaminergic neurons. To address this question, Ahfeldt et al.123 used hMLO to study the roles of 3 severe PD-associated mutations mentioned above (in PARKIN/PRKN, DJ1/PARK7, and ATP13A2/PARK9) through genomic editing of a healthy control iPSC line. Increased levels of oxidative stress are found in all PD lines. Increased death of DNs upon differentiation was found only in the PARKIN knockout line. Using quantitative proteomics, they observed dysregulation of mitochondrial and lysosomal function in all of the lines, as well as common and distinct molecular defects caused by the different PD genes. Dysregulation of PD-relevant pathways indicate shared and distinct molecular signatures among the isogenic PD cell lines. Identified shared or specific dysregulated candidate genes may inform efforts to find therapeutic targets and to stratify PD pathology and patients on a molecular level. Besides, Parfitt et al.124 focused on the role of DJ1 in an iPSC-derived midbrain organoid model deficient for DJ1 activity, which illustrated how disruption in lysosomal proteolysis led by astrocytes contributes to the accumulation of advanced glycation end products (AGEs) and increased α-syn phosphorylation. This study emphasized the importance of astrocytes in maintaining proteostasis within the neuronal environment, particularly highlighting their role in reversing proteolysis deficits in DJ1 knockout midbrain neurons. In another study125, midbrain organoids were used as a tool to help researchers identify a unique subtype of neurons, characterized by the expression of the Parkinson’s disease risk gene RIT2.
Construct hMLOs using PD causative exogenous stimuli
In general, the hMLO models of PD are mainly induced by iPSCs carrying PD-related mutations, with downstream experiments principally study the pathogenic molecular mechanism of mutation-related PD. However, similar to the establishment of animal model of PD, the induction of PD hMLOs can also use neurotoxins. Compared with gene-based methods, toxin-based method has its irreplaceable advantage: the mechanisms of these neurotoxic drugs in PD pathogenesis can be comprehensively studied. So far, two successful in-depth cases have been reported, use MPTP and 6-OHDA as exogenous neurotoxic stimuli, respectively46,126.
To replicate the neuropathology of PD, many previous studies had used animal models with a variety of dopaminergic toxins127. Among them, MPTP is known to be the most reliable and frequently used toxin due to its ability to stably induce clinical symptoms that are indistinguishable from PD128,129. After crossing the blood-brain barrier, MPTP is converted first into 1-methyl-4-phenyl-2,3-dihydropyridinium (MPDP) by monoamine oxidase B (MAO-B) and then into 1-methyl-4-phenylpyridinium (MPP+) in astrocytes129. Though astrocytes play a key role in the mode of action of MPTP129,130, MPTP, a representative and reliable dopaminergic neurotoxin, has not been properly used in previous iPSC-based in vitro modeling studies of PD. Kwak et al.46 generated a special type of hMLO named DAC3.0 MOs which contain a large number of glial cells, such as astrocyte. MPTP-treated DAC3.0 Mos exhibited massive cell death, and the number of apoptotic cells increased in a dose-dependent manner, indicating that the cell death induced in DAC3.0 MOs is mediated solely by MPTP treatment. Notably, MPTP-mediated cell death is observed largely in TH-positive mDA neurons but rarely in other cellular components (GABAergic neurons, astrocytes, and oligodendrocytes etc.) Taking the information together, their data indicates that DAC3.0 MOs produced by their modified protocol contain functional glial cells that facilitate the action of MPTP, a representative dopaminergic neurotoxin, which could be used for the in vitro modeling of PD.
Another similar research is done by Monzel et al.126. They developed a neurotoxin-induced PD organoid model by neurotoxic compound 6-OHDA treatment. Cell quantification by flow cytometry revealed that exposure to 6-OHDA caused a concentration-dependent reduction in the amount of living DANs. 6-OHDA treatment leads to a decrease in the amount of TH+ cells and to neurite fragmentation, in other words, it led to a decrease in the complexity of DANs and increase of fragmented neurites. Results showed that it is a valuable tool for advanced in vitro PD modeling.
Using hMLOs for appropriate PD toxicity predictions
Approximately 90% of cases are of sporadic and idiopathic origin131,132, and many studies hypothesize that genetic and environmental factors, including exposure to toxicants, may play a role in the long-term etiology of PD133,134,135. However, Parkinsonism in humans can also have more direct causes, where specific compounds can selectively and acutely ablate dopaminergic neurons. For example, MPTP, a toxic byproduct of drug synthesis, caused PD-like symptoms in illicit drug users that had unintentionally exposed themselves to the compound136,137. Following the discovery of MPTP’s effects, similar specific toxicities were identified for other compounds, including the common pesticides rotenone and paraquat138,139. Furthermore, many therapeutic drugs are known to cause drug-induced Parkinsonism (DIP) in patients (the second most common form of Parkinsonism in aged patients after PD)139,140,141. While DIP is often reversible as it is mostly caused by the blockage of the dopamine receptor at the post-synapse141, some drugs, including the antipsychotic haloperidol, may also have additional neurotoxic effects and thus directly damage dopaminergic neurons142,143,144. Human data on the molecular details of these side effects are scarce due to limited access to tissue samples. Toxicity testing is a crucial step in the development and approval of chemical compounds for human contact and consumption. However, existing model systems often fall short in their prediction of human toxicity in vivo because they may not sufficiently recapitulate human physiology. The complexity of 3D human organ-like cell culture systems (“organoids”) can generate potentially more relevant models of human physiology and disease, including toxicity predictions. As previously described in section “Construct hMLOs using PD causative exogenous stimuli”, neurotoxic drugs, including MPTP and 6-OHDA, can be used for the construction of hMLOs with PD phenotypes. Compared with the mainstream gene-based models, toxin-based models can be further used as an ideal platform for the further study of PD pathogenesis caused by these neurotoxins. Followed by advanced technics such as microscopy-based phenotyping in a high-content fashion, neurotoxic effect on dopaminergic neurons can be assessed123.
Oxidative stress is a crucial causative factor for PD progression145, which is caused by an imbalance between the generation and detoxification of reactive oxygen and nitrogen species (ROS/RNS). This imbalance plays an important role in brain aging and age-related neurodegenerative diseases. In the context of PD, the sensitivity of dopaminergic neurons in the substantia nigra pars compacta to oxidative stress is considered a key factor of PD pathogenesis. David et al.146 studied the effect of different oxidative stress-inducing compounds (6-OHDA, MPTP or MPP+) on the population of dopaminergic neurons in an iPSC-derived hMLO model. Treatment with 6-OHDA, MPTP or MPP+ at 4 weeks of differentiation disrupted the dopaminergic neuronal phenotype in hMLOs. 6-OHDA increased ROS production and decreased mitochondrial function most efficiently. It further induced the greatest changes in gene expression and metabolites related to oxidative stress and mitochondrial dysfunction. Co-culturing hMLOs with an endothelial barrier using a transwell system allowed the assessment of differential penetration capacities of the tested compounds and the damage they caused in the dopaminergic neurons within the hMLOs. In conclusion, treatment with compounds known to induce PD-like phenotypes in vivo caused molecular deficits and loss of dopaminergic neurons in the hMLOs model. This approach therefore recapitulates common animal models of neurodegenerative processes in PD at similarly high doses.
So far, the inherent biological heterogeneity and cumbersome generation and analysis of organoids has rendered efficient, unbiased, high throughput evaluation of toxic effects in these systems challenging. Recent advances in both standardization and quantitative fluorescence imaging enabled researchers to dissect the toxicities of compound exposure to separate cellular subpopulations within human organoids at the single-cell level in a framework that is compatible with high throughput approaches147,148. Renner et al.147 screened a library of 84 compounds in standardized human automated midbrain organoids (AMOs) generated from two independent cell lines correctly recognized known nigrostriatal toxicants. This approach further identified the flame retardant 3,3’,5,5’-tetrabromobisphenol A (TBBPA) as a selective toxicant for dopaminergic neurons in the context of human midbrain-like tissues for the first time. Further, 3D AMOs demonstrate higher sensitivity in than in 2D cultures to the known neurotoxic effects of the pesticide lindane, which proved the feasibility of quantitatively assessing cell-type-specific toxicity in human organoids in vitro.
Using hMLOs for appropriate PD treatment
At present, the mainstream treatment of PD is drug therapy such as levodopa and surgeries like deep brain stimulation149,150,151. These treatment options were designed to relieve the symptoms of PD or mitigate side effects of antiparkinsonian drugs rather than intervene in the underlying disease process. To achieve meaningful progress in halting or delaying the progression of cell loss in PD, a greater understanding of the disease process is needed. As an optimized in vitro model, the appearance of hMLOs may provide a new perspective for PD treatment152. We have compiled in Table 2 the recent advancements in using hMLOs for the therapeutic applications targeting PD.
PD treatment using neural stem cells derived from hMLOs
Recent studies have demonstrated that stem cell-derived mDA neurons or mDA progenitors could indeed functionally integrate into striatonigral circuits153, and provide some symptomatic relief in a non-human primate model of PD without forming tumors154. The extent of recovery in the neurologic score and spontaneous movement of PD monkeys grafted with iPSC-derived cells was similar or lower than those produced by L-DOPA drug administration154. The outcome of PD clinical trials cannot be predicted with the information currently available, but studies to improve cell therapeutic efficacy and safety should not be abandoned but continued until a complete success in the clinical transition is reached. Therefore, as organoids have been shown to efficiently integrate into rodent neural circuits after transplantation155, using dopamine-producing hMLOs may prove to be a useful development for therapeutic purposes. In this regard, the following issues in donor cell preparation need to be further addressed for a successful clinical translation of PD cell therapy, which are, the high expression of midbrain factors, the existence of astrocyte differentiation, and the stability and reproducibility of hMLOs.
Successful clinical translation of stem cell-based therapy largely relies on the scalable and reproducible preparation of donor cells with potent therapeutic capacities. Kim et al.55 use hMLOs to prepare cells for PD therapy. In this study, neural stem cells (NSCs) were isolated from midbrain organoids (Og-NSCs), which expanded stably and differentiated into mDA neurons. Unprecedentedly high proportion of cells expressed midbrain-specific factors, with relatively low cell line and batch-to-batch variations. Single cell transcriptome analysis followed by in vitro assays indicated that the majority of cells in the Og-NSC cultures are ventral midbrain (VM)-patterned with low levels of cellular senescence and mitochondrial stress, compared to those derived from 2D-culture environments. Notably, compared to traditional hMLO that aims to mimic the whole midbrain area, the VM- patterned hMLOs are prone to produce more A9 DA neurons, which is essential in PD development molecular profile55,156,157,158. Moreover, in contrast to current methods yielding mDA neurons without astrocyte differentiation, mDA neurons that differentiated from Og-NSCs were interspersed with astrocytes as in the physiologic brain environment. Thus, the Og-NSC-derived mDA neurons exhibited improved synaptic maturity, functionality, resistance to toxic insults, and faithful expressions of the midbrain-specific factors. They further transplant Og-NSCs into 6-OHDA-lesioned hemi-parkinsonian rats to testify their therapeutic effect, the results showed reproducible behavioral restoration and mDA neuron engraftment in PD animals transplanted with Og-NSCs. Though this experiment broadens our horizon in PD treatment using hMLO-derived NSCs, only the H9 cell line is used for PD treatment, for a wide range of clinical utility of the Og-NSC method, more conclusive results should be verified in a variety of hESC/iPSC lines. Zheng et al.159 explored the potential of hMLOs derived from iPSC for treating PD. Their study found that hMLOs, particularly at Day 15 of differentiation, are effective in replenishing dopaminergic neurons when transplanted into PD mouse models. Post-transplantation, these cells demonstrated survival, maturation, and integration into the host brain, effectively restoring motor functions and establishing bidirectional connections with natural brain targets, without tumor formation.
Using hMLOs for PD drug selection
The road to discover novel therapeutics for neurological disorders including PD has been severely hampered by the lack of access to relevant testing platforms. One of the reasons responsible for this low success rate is that conventional 2D cell culture models are not accurate enough predictors of how drugs will work in humans. 3D brain organoids differentiated from iPSCs to resemble specific parts of the human brain, which include architecture composition and physiology, can provide an alternative system that may lead to breakthroughs in key areas of drug testing and toxicological evaluation. Having reliable and scalable iPSC-derived brain organoid models that can much more accurately predict human drug responses will significantly increase success rate in developing treatments for brain-related disorders. In this background, organoids constitute a relevant platform to identify novel therapeutic compounds and to assess their efficacy on specific phenotypes. For example, Kim et al.31 showed that α-synuclein accumulation could be reduced in LRRK2 G2019S hMLO through treatment with a LRRK2 kinase activity inhibitor (GSK2578215A), but also by knocking down the expression of TXNIP, which their study had identified as a central mediator of G2019S pathology. Jarazo et al.160 also found that treatment with the HP-ß-CD compound improved mDA neuronal differentiation in PINK1 and PRKN-mutated hMLO, likely through increased mitophagy. Besides, Kim et al.161 developed an optogenetics-assisted system, OASIS, which rapidly induces α-synuclein aggregates in PD iPSC-derived midbrain neurons and organoids. This method identified BAG956, a compound that effectively reverses PD phenotypes by enhancing autophagic clearance of pathological α-syn aggregates. Recently, Shin et al.162 introduced a novel biohybrid robot-on-a-chip incorporating a brain organoid, motor neuron spheroids, and muscle bundle for evaluating drug effects on neurodegenerative diseases like PD. This system uniquely integrates a patient-derived midbrain organoid to measure muscle movement, demonstrating significant improvement in muscle bundle movement in response to levodopa. This model overcomes previous limitations by successfully mimicking human motor system functions, providing a promising tool for PD drug evaluation.
Up to now, the two main approaches for drug discovery are target-based screenings, where the modulation of the activity of a previously identified druggable target is used as readout, and phenotypic screenings, in which the rescue of a phenotype in a disease model (cells, tissue, animal etc.) serves as readout163,164. In the past four decades, the majority of investment for drug discovery was biased towards target-based screenings164. These highly standardized large-scale assays can be performed in classical cell lines or even with isolated proteins in vitro. However, despite increasing investment, drug discovery has a high failure rate especially when it comes to neurodegenerative diseases (NDD)165. In fact, there are still no disease modifying drugs approved for the major NDD, Alzheimer’s disease (AD) and PD165. In the last 20 years only 5 first-in-class small molecule drugs for NDD were approved, of which none was discovered in target-based screenings but in phenotypic screenings166. Multifactorial diseases like PD are only poorly represented in a screening focusing on a single target molecule, as the “one-drug-one-target” approach does not take into account the complexity of such diseases167. Patient-derived hMLO models become an increasingly powerful tool to model human diseases for precision medicine approaches. The identification of robust cellular disease phenotypes in these models paved the way towards high throughput screenings (HTS) including the implementation of laboratory advanced automation. Recently, researchers describe an integrated, complex automated platform for HTS in translational research setting168. The comprehensive integrated automated platform can be used for designed to maintain and expand different cell types (adherent and suspension cells), differentiate iPSC-based hMLOs and to perform high throughput and HTS. This allows for produce sufficient cells for HTS/HCS purposes and can be adapted to meet special requirements of other cell types enabling novel precision medicine approaches across diseases. However, due to the needs of high-throughput, high content and small variations between batches during HST, there is a long way to go for adequate combination of these two prevalence techniques169,170,171.
Discussion
Parkinson’s disease, by its progressive and irreversible nature, has been challenging for clinicians to diagnose and prognosticate. Reliable markers of early disease have yet to be identified and brain tissue biopsies rarely provide enough benefits to outweigh the risks of the procedure16. Besides, there are still no disease-modifying drugs available to prevent or reverse these neurodegenerative phenomena. One of the existing barriers to potential therapy development is the remarkably unclear understood nature of disease development142. Similarly, scalability and ethical barriers limit the high-throughput drug-testing potential of the established animal models. The emergence of iPSCs technology brought light to mimicking the age-dependent neurodegenerative diseases such as PD, AD, and amyotrophic lateral sclerosis (ALS). Researchers have been exploring the potential of iPSC-derived cells for disease modeling, drug screening, and cell replacement therapy in these disorders. The advent of organoids has elevated the application of iPSC-based technology to new heights. It presents a novel model for PD simulation, disease progression prediction, drug screening, and therapeutic interventions55.
Despite significant progress in modeling neurodegenerative diseases such as PD with brain organoids derived from stem cells, challenges remain in fully replicating the regional specificity and functionality of the human brain. A notable challenge is the inadequacy of vascularization, which affects not just the traditional role of vessels in supplying nutrients and oxygen, but also key aspects of disease modeling172. Vascularization is crucial for supporting the survival of cells at the center of organoids, preventing apoptosis due to insufficient nutrients and oxygen, and allowing the organoids to grow to a larger size, which is necessary for long-term studies of PD progression and intervention measures. Babu et al.173 provided a detailed overview of the relevant research progress. Moreover, the integrity of the blood-brain barrier (BBB) is compromised in PD174, and its effective simulation is essential for understanding its pathological role in PD, such as changes during neuroinflammation and neurodegenerative processes175, and for assessing disease impact and treatment responses. Therefore, a realistic reproduction of the BBB in organoid models is critically important for in-depth study of these processes. Lastly, simulating the mechanism of drug delivery through the BBB is crucial for PD treatment strategies. The vascularization of organoid models provides a new method for studying how to improve the efficiency of drug delivery and assess the penetration capabilities of new drugs, especially considering the unique permeability characteristics of the BBB in PD patients176. To address the issue of vascularization in brain organoids, scientists have adopted a variety of strategies. These strategies include the use of co-culturing techniques with cells, the introduction of specific growth factors, and the development of new bioengineering methods aimed at enhancing the capability and functionality of vascular formation in organoid models155,177,178. Dao et al.179 successfully developed the world’s first human mini-brain that includes a fully functional BBB. These assembloids effectively recapitulated the complex anatomy and BBB breakdown seen in patients with cerebral cavernous malformations (CCMs), providing insights into disease mechanisms and potential therapeutic targets. Addressing these challenges not only enhances the biological relevance and research efficiency of organoid models but also advances the simulation and treatment research of complex neurological diseases like PD. Another issue should be taken into account is the lack of immune cell recruitment95,180. As immune cells participate in many neurodegenerative diseases, the role of immune cells should never be ignored. Future protocols should aim to that allow the growth of immune cells, especially microglia, in existing organoids181. As they regulate synapse formation and assist in the generation and maintenance of neuronal circuits in the human brain182. Due to lack of immune cells and vasculature, the ability of brain organoids to replicate the human brain remains less. Therefore, the recruitment of immune cells and vascular networks within organoids will expand the applicability of these systems to study more complex phenomena such as defined cell-cell interactions, circuit functionality, and later developmental events such as myelination and plasticity183. Recent study has reported a method for integrating microglia into iPSC-derived midbrain organoids, this midbrain-microglia assembloids shed light on the new model for neuroinflammation in Parkinson’s disease184. Furthermore, although the advantages of 3D organoids over traditional 2D culture are obvious as mentioned above, the 2D approach has its own gains. The heterogeneity of 2D cultured cells between different batches was relatively low, with high reproducibility. Besides, 2D environment is quite convenient to use regarding the culture condition requirements, cell growth potential, flexible intervention, and possibility of close microscopic observation185. Moreover, 3D culture is not superior to 2D culture in all condition. Given the higher interstitial edema and tissue necrosis, which are still common in conventional induction methods, it has been reported that 3D culturation may not perform as well as that in 2D conditions186.
In the long run, the application of hMLOs should be further explored in vary aspects, including the construction of muti-area brain organoids, the direct transplantation of organoids and the application of personalized medication. (1) The construction of area-fused brain organoids. The human central nervous system (CNS) develops from several distinct vesicles into multiple intertwined regions. During this process, a range of migratory streams arise where progenitors generated in one place migrate and integrate into other areas187,188,189,190, and complex networks emerge, neurons branching and projecting across multiple regions191. Yet convoluted, the mechanism underpinning the formation of the human CNS is a highly ordered process that needs to be understood. Many diseases are the result of barriers of projection, migration or neuronal interaction between different brain regions. Although some organizing centers were observed in brain organoids192, which are critical for regional brain patterning, most of the spatial identities in organoids appeared in an uncontrolled manner, thereby limiting the study of complex interregional interactions. Application of patterning factors and/or chemicals allows us to pattern brain organoids into different brain regions193,194,195,196, which provides researchers “LEGO blocks” to establish fused organoid approaches. The development of fused organoids, also called assembloids197, opens a new avenue to investigate interregional dynamics in the embryonic brain. Fusing two brain organoids pre-patterned into different regional identities enables the study of interregional interactions. Organoids made by the fusion approach can help to deconstruct organogenesis by reconstructing the brain piece by piece. The development of PD is the result of abnormal projection from substantia nigra into the striatum, the application of fusion organoids can help to fully understand the pathogenesis of PD in closed neural circuits. (2) The direct transplantation of hMLOs. In a recent study, an in vivo model of vascularized human brain organoids was developed by transplanting organoids into the superficial cortex of mice155. Although widespread axonal extension outside the graft area was observed, region-specific long projections were not reported. Previously, reported methods produced cerebral organoids containing multiple lumens or neural tubes37,198, which makes them difficult to use for transplantation therapy. In addition, large organoids might cause more damage to recipients than small organoids if they are transplanted in deep areas of the brain. It is possible that small brain organoids can alleviate these safety concerns and are more amenable to injection into deep brain regions. Liu et al. optimized a culturing protocol capable of efficiently generating small human cerebral organoids199. After transplantation into the mouse medial prefrontal cortex, the grafted human cerebral organoids survived and extended long projections to basal brain regions. Importantly, the grafted human cerebral organoids functionally integrated into pre-existing neural circuits by forming bidirectional synaptic connections with the mouse host neurons. Besides, mouse received the grafted organoids present with potentiation of the startle fear response. This study showed that subcortical projections can be established by microtransplantation and may provide crucial insights into the therapeutic potential of hMLOs for PD treatment. Recent studies also report homotopic transplantation of 2D-drived neuron into PD rat brain combined with AAV-mediate GNDF injection, the grafted mDA in SN showed a better connection with host striatum than the traditional ectopic method200. Together with a study showing that displaced VM-progenitors with α-synuclein triplication into the brain of PD rats can aggravate the disease phenotype201, these studies hints that direct homotopic transplantation of hMLOs may be a promising way, which can be used to explore the pathogenicity of PD by transplanting mutation-specific organoids into normal rodent models or the therapeutic effect of by grafting healthy organoids into PD-phenotypic ones. (3) In recent years, other emerging technologies such as snRNA-seq202,203, spatial transcriptomics203, 3D bioprinting204, preformed fibrils205, and fetal brain in vitro self-organization into organoids206 have been incorporated into research on brain-like organs, offering promising avenues for further exploration in understanding PD. These innovative techniques open up new dimensions for studying PD and hold significant potential for advancement in this field.
This paper reviews the development of hMLOs since their first appearance and systematically describes their applications in PD model construction, toxicity prediction and treatment so far. Until now, there are still some defects in the induction methods, such as lack of vascularization and immunity. Meanwhile, the application of hMLOs also needs to be further explored. The construction of complex organoids with multiple brain regions and the direct transplantation of small organoids may be a promising development direction in the future. With the continuous in-depth exploration of hMLOs, researchers have made thrilling steps in their methodology and application. For the foreseeable future, hMLOs may play an increasingly important role in the mechanism research and treatment of PD.
References
-
Fearnley, J. M. & Lees, A. J. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain 114(Pt 5), 2283–2301 (1991).
-
GBD 2016 Neurology Collaborators. Global, regional, and national burden of neurological disorders, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 18, 459–480 (2019).
-
Dauer, W. & Przedborski, S. Parkinson’s disease: mechanisms and models. Neuron 39, 889–909 (2003).
-
Kalia, L. V. & Lang, A. E. Parkinson’s disease. Lancet 386, 896–912 (2015).
-
Bloem, B. R., Okun, M. S. & Klein, C. Parkinson’s disease. Lancet 397, 2284–2303 (2021).
-
Obeso, J. A. et al. Past, present, and future of Parkinson’s disease: a special essay on the 200th Anniversary of the Shaking Palsy. Mov. Disord. 32, 1264–1310 (2017).
-
Jankovic, J. Parkinson’s disease: clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 79, 368–376 (2008).
-
Schapira, A. H. V., Chaudhuri, K. R. & Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 18, 435–450 (2017).
-
Schapira, A. H. V., Chaudhuri, K. R. & Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 18, 509 (2017).
-
Sulzer, D. & Surmeier, D. J. Neuronal vulnerability, pathogenesis, and Parkinson’s disease. Mov. Disord. 28, 41–50 (2013).
-
Chandra, R., Hiniker, A., Kuo, Y.-M., Nussbaum, R. L. & Liddle, R. A. α-Synuclein in gut endocrine cells and its implications for Parkinson’s disease. JCI Insight 2, e92295 (2017).
-
Spillantini, M. G. et al. Α-synuclein in Lewy bodies. Nature 388, 839–840 (1997).
-
Wakabayashi, K., Hayashi, S., Yoshimoto, M., Kudo, H. & Takahashi, H. NACP/α-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson’s disease brains. Acta Neuropathol. 99, 14–20 (2000).
-
Michel, P. P., Hirsch, E. C. & Hunot, S. Understanding dopaminergic cell death pathways in Parkinson disease. Neuron 90, 675–691 (2016).
-
Zhang, G. et al. New perspectives on roles of Α-synuclein in Parkinson’s disease. Front. Aging Neurosci. 10, 370 (2018).
-
Di Lullo, E. & Kriegstein, A. R. The use of brain organoids to investigate neural development and disease. Nat. Rev. Neurosci. 18, 573–584 (2017).
-
Zeng, X.-S., Geng, W.-S. & Jia, J.-J. Neurotoxin-induced animal models of Parkinson disease: pathogenic mechanism and assessment. ASN Neuro 10, 1759091418777438 (2018).
-
Chia, S. J., Tan, E.-K. & Chao, Y.-X. Historical perspective: models of Parkinson’s disease. Int. J. Mol. Sci. 21, 2464 (2020).
-
Gasser, T. Molecular pathogenesis of Parkinson disease: insights from genetic studies. Expert Rev. Mol. Med. 11, e22 (2009).
-
Taguchi, T., Ikuno, M., Yamakado, H. & Takahashi, R. Animal model for prodromal Parkinson’s disease. Int. J. Mol. Sci. 21, 1961 (2020).
-
Hartung, T. Thoughts on limitations of animal models. Parkinsonism Relat. Disord. 14(Suppl 2), S81–S83 (2008).
-
Kokjohn, T. A. & Roher, A. E. Amyloid precursor protein transgenic mouse models and Alzheimer’s disease: understanding the paradigms, limitations, and contributions. Alzheimers Dement. 5, 340–347 (2009).
-
Takahashi, K. & Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 (2006).
-
Miller, J. D. et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 13, 691–705 (2013).
-
Kriks, S. et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 480, 547–551 (2011).
-
Nolbrant, S., Heuer, A., Parmar, M. & Kirkeby, A. Generation of high-purity human ventral midbrain dopaminergic progenitors for in vitro maturation and intracerebral transplantation. Nat. Protoc. 12, 1962–1979 (2017).
-
Kirkeby, A. et al. Predictive markers guide differentiation to improve graft outcome in clinical translation of hESC-based therapy for Parkinson’s disease. Cell Stem Cell 20, 135–148 (2017).
-
Sulzer, D. et al. Neuromelanin biosynthesis is driven by excess cytosolic catecholamines not accumulated by synaptic vesicles. Proc. Natl Acad. Sci. USA 97, 11869–11874 (2000).
-
Zecca, L., Zucca, F. A., Wilms, H. & Sulzer, D. Neuromelanin of the substantia nigra: a neuronal black hole with protective and toxic characteristics. Trends Neurosci. 26, 578–580 (2003).
-
Pașca, S. P. The rise of three-dimensional human brain cultures. Nature 553, 437–445 (2018).
-
Kim, H. et al. Modeling G2019S-LRRK2 sporadic Parkinson’s disease in 3D midbrain organoids. Stem Cell Rep. 12, 518–531 (2019).
-
Ren, W. et al. Single Lgr5- or Lgr6-expressing taste stem/progenitor cells generate taste bud cells ex vivo. Proc. Natl Acad. Sci. USA 111, 16401–16406 (2014).
-
Xia, Y. et al. The generation of kidney organoids by differentiation of human pluripotent cells to ureteric bud progenitor-like cells. Nat. Protoc. 9, 2693–2704 (2014).
-
Rookmaaker, M. B., Schutgens, F., Verhaar, M. C. & Clevers, H. Development and application of human adult stem or progenitor cell organoids. Nat. Rev. Nephrol. 11, 546–554 (2015).
-
Adams, J. W., Cugola, F. R. & Muotri, A. R. Brain organoids as tools for modeling human neurodevelopmental disorders. Physiology 34, 365–375 (2019).
-
Kim, S.-H. & Chang, M.-Y. Application of human brain organoids-opportunities and challenges in modeling human brain development and neurodevelopmental diseases. Int. J. Mol. Sci. 24, 12528 (2023).
-
Lancaster, M. A. et al. Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379 (2013).
-
Muguruma, K., Nishiyama, A., Kawakami, H., Hashimoto, K. & Sasai, Y. Self-organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells. Cell Rep. 10, 537–550 (2015).
-
Qian, X. et al. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell 165, 1238–1254 (2016).
-
Lancaster, M. A. & Knoblich, J. A. Organogenesis in a dish: modeling development and disease using organoid technologies. Science 345, 1247125 (2014).
-
Quadrato, G. et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature 545, 48–53 (2017).
-
Pamies, D. et al. A human brain microphysiological system derived from induced pluripotent stem cells to study neurological diseases and toxicity. ALTEX 34, 362–376 (2017).
-
Tieng, V. et al. Engineering of midbrain organoids containing long-lived dopaminergic neurons. Stem Cells Dev. 23, 1535–1547 (2014).
-
Jo, J. et al. Midbrain-like organoids from human pluripotent stem cells contain functional dopaminergic and neuromelanin-producing neurons. Cell Stem Cell 19, 248–257 (2016).
-
Monzel, A. S. et al. Derivation of human midbrain-specific organoids from neuroepithelial stem cells. Stem Cell Rep. 8, 1144–1154 (2017).
-
Kwak, T. H. et al. Generation of homogeneous midbrain organoids with in vivo-like cellular composition facilitates neurotoxin-based Parkinson’s disease modeling. Stem Cells 38, 727–740 (2020).
-
La Manno, G. et al. Molecular diversity of midbrain development in mouse, human, and stem cells. Cell 167, 566–580.e19 (2016).
-
Tiklová, K. et al. Single-cell RNA sequencing reveals midbrain dopamine neuron diversity emerging during mouse brain development. Nat. Commun. 10, 581 (2019).
-
Liu, G. et al. Aldehyde dehydrogenase 1 defines and protects a nigrostriatal dopaminergic neuron subpopulation. J. Clin. Invest. 124, 3032–3046 (2014).
-
Xiong, M. et al. Human stem cell-derived neurons repair circuits and restore neural function. Cell Stem Cell 28, 112–126.e6 (2021).
-
Burns, T. C., Li, M. D., Mehta, S., Awad, A. J. & Morgan, A. A. Mouse models rarely mimic the transcriptome of human neurodegenerative diseases: a systematic bioinformatics-based critique of preclinical models. Eur. J. Pharm. 759, 101–117 (2015).
-
Bolognin, S. et al. 3D cultures of Parkinson’s disease-specific dopaminergic neurons for high content phenotyping and drug testing. Adv. Sci. 6, 1800927 (2019).
-
Klima, S. et al. A human stem cell-derived test system for agents modifying neuronal N-methyl-D-aspartate-type glutamate receptor Ca2+-signalling. Arch. Toxicol. 95, 1703–1722 (2021).
-
Zanetti, C. et al. Monitoring the neurotransmitter release of human midbrain organoids using a redox cycling microsensor as a novel tool for personalized Parkinson’s disease modelling and drug screening. Analyst 146, 2358–2367 (2021).
-
Kim, S. W. et al. Neural stem cells derived from human midbrain organoids as a stable source for treating Parkinson’s disease: midbrain organoid-NSCs (Og-NSC) as a stable source for PD treatment. Prog. Neurobiol. 204, 102086 (2021).
-
Lee, H.-K. et al. Three dimensional human neuro-spheroid model of Alzheimer’s disease based on differentiated induced pluripotent stem cells. PLoS ONE 11, e0163072 (2016).
-
Yan, Y. et al. Modeling neurodegenerative microenvironment using cortical organoids derived from human stem cells. Tissue Eng. Part A 24, 1125–1137 (2018).
-
Osaki, T., Uzel, S. G. M. & Kamm, R. D. Microphysiological 3D model of amyotrophic lateral sclerosis (ALS) from human iPS-derived muscle cells and optogenetic motor neurons. Sci. Adv. 4, eaat5847 (2018).
-
Conforti, P. et al. Faulty neuronal determination and cell polarization are reverted by modulating HD early phenotypes. Proc. Natl Acad. Sci. USA 115, E762–E771 (2018).
-
Hor, J. H. et al. Cell cycle inhibitors protect motor neurons in an organoid model of Spinal Muscular Atrophy. Cell Death Dis. 9, 1100 (2018).
-
Blauwendraat, C., Nalls, M. A. & Singleton, A. B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 19, 170–178 (2020).
-
Nalls, M. A. et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102 (2019).
-
Senkevich, K. & Gan-Or, Z. Autophagy lysosomal pathway dysfunction in Parkinson’s disease; evidence from human genetics. Parkinsonism Relat. Disord. 73, 60–71 (2020).
-
Balestrino, R. & Schapira, A. H. V. Parkinson disease. Eur. J. Neurol. 27, 27–42 (2020).
-
Hogberg, H. T. et al. Toward a 3D model of human brain development for studying gene/environment interactions. Stem Cell Res Ther. 4(Suppl 1), S4 (2013).
-
Kirkeby, A. et al. Generation of regionally specified neural progenitors and functional neurons from human embryonic stem cells under defined conditions. Cell Rep. 1, 703–714 (2012).
-
Polymeropoulos, M. H. et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047 (1997).
-
Fujioka, S. et al. Update on novel familial forms of Parkinson’s disease and multiple system atrophy. Parkinsonism Relat. Disord. 20(Suppl 1), S29–S34 (2014).
-
Kasten, M. & Klein, C. The many faces of α-synuclein mutations. Mov. Disord. 28, 697–701 (2013).
-
Krüger, R. et al. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat. Genet. 18, 106–108 (1998).
-
Lesage, S. et al. G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 73, 459–471 (2013).
-
Zarranz, J. J. et al. The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173 (2004).
-
Chartier-Harlin, M.-C. et al. Α-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364, 1167–1169 (2004).
-
Farrer, M. et al. Comparison of kindreds with parkinsonism and α-synuclein genomic multiplications. Ann. Neurol. 55, 174–179 (2004).
-
Nishioka, K. et al. Clinical heterogeneity of α-synuclein gene duplication in Parkinson’s disease. Ann. Neurol. 59, 298–309 (2006).
-
Ibáñez, P. et al. Causal relation between α-synuclein gene duplication and familial Parkinson’s disease. Lancet 364, 1169–1171 (2004).
-
Muenter, M. D. et al. Hereditary form of parkinsonism-dementia. Ann. Neurol. 43, 768–781 (1998).
-
Book, A. et al. A meta-analysis of α-synuclein multiplication in familial parkinsonism. Front. Neurol. 9, 1021 (2018).
-
Delenclos, M. et al. Cellular models of α-synuclein toxicity and aggregation. J. Neurochem. 150, 566–576 (2019).
-
Jo, J. et al. Lewy body-like inclusions in human midbrain organoids carrying glucocerebrosidase and α-synuclein mutations. Ann. Neurol. 90, 490–505 (2021).
-
Mohamed, N.-V. et al. Midbrain organoids with an SNCA gene triplication model key features of synucleinopathy. Brain Commun. 3, fcab223 (2021).
-
Becerra-Calixto, A. et al. Lewy body-like pathology and loss of dopaminergic neurons in midbrain organoids derived from familial Parkinson’s disease patient. Cells 12, 625 (2023).
-
Muwanigwa, M. N. et al. Α-synuclein pathology is associated with astrocyte senescence in a midbrain organoid model of familial Parkinson’s disease. Mol. Cell Neurosci. 128, 103919 (2024).
-
Di Fonzo, A. et al. A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson’s disease. Lancet 365, 412–415 (2005).
-
Paisán-Ruíz, C. et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44, 595–600 (2004).
-
Daher, J. P. L. et al. Leucine-rich Repeat Kinase 2 (LRRK2) Pharmacological inhibition abates α-synuclein gene-induced neurodegeneration. J. Biol. Chem. 290, 19433–19444 (2015).
-
Hsieh, C.-H. et al. Functional impairment in miro degradation and mitophagy is a shared feature in familial and sporadic Parkinson’s disease. Cell Stem Cell 19, 709–724 (2016).
-
Lin, X. et al. Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant α-synuclein. Neuron 64, 807–827 (2009).
-
Manzoni, C. & Lewis, P. A. Dysfunction of the autophagy/lysosomal degradation pathway is a shared feature of the genetic synucleinopathies. FASEB J. 27, 3424–3429 (2013).
-
Chesselet, M.-F., Fleming, S., Mortazavi, F. & Meurers, B. Strengths and limitations of genetic mouse models of Parkinson’s disease. Parkinsonism Relat. Disord. 14(Suppl 2), S84–S87 (2008).
-
Giasson, B. I. et al. Neuronal α-synucleinopathy with severe movement disorder in mice expressing A53T human α-synuclein. Neuron 34, 521–533 (2002).
-
Lee, M. K. et al. Human α-synuclein-harboring familial Parkinson’s disease-linked Ala-53 -> Thr mutation causes neurodegenerative disease with α-synuclein aggregation in transgenic mice. Proc. Natl Acad. Sci. USA 99, 8968–8973 (2002).
-
Masliah, E. et al. Dopaminergic loss and inclusion body formation in α-synuclein mice: implications for neurodegenerative disorders. Science 287, 1265–1269 (2000).
-
Beal, M. F. Experimental models of Parkinson’s disease. Nat. Rev. Neurosci. 2, 325–334 (2001).
-
Chung, C. Y. et al. Identification and rescue of α-synuclein toxicity in Parkinson patient-derived neurons. Science 342, 983–987 (2013).
-
Kluss, J. H., Mamais, A. & Cookson, M. R. LRRK2 links genetic and sporadic Parkinson’s disease. Biochem. Soc. Trans. 47, 651–661 (2019).
-
Smits, L. M. et al. Modeling Parkinson’s disease in midbrain-like organoids. NPJ Parkinsons Dis. 5, 5 (2019).
-
Su, C.-J. et al. Thioredoxin-interacting protein induced α-synuclein accumulation via inhibition of autophagic flux: Implications for Parkinson’s disease. CNS Neurosci. Ther. 23, 717–723 (2017).
-
Zagare, A. et al. Midbrain organoids mimic early embryonic neurodevelopment and recapitulate LRRK2-p.Gly2019Ser-associated gene expression. Am. J. Hum. Genet. 109, 311–327 (2022).
-
Valente, E. M. et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160 (2004).
-
Gandhi, S. et al. PINK1 protein in normal human brain and Parkinson’s disease. Brain 129, 1720–1731 (2006).
-
Hoepken, H.-H. et al. Mitochondrial dysfunction, peroxidation damage and changes in glutathione metabolism in PARK6. Neurobiol. Dis. 25, 401–411 (2007).
-
Deas, E. et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 20, 867–879 (2011).
-
Okatsu, K. et al. PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun. 3, 1016 (2012).
-
Matsuda, S., Kitagishi, Y. & Kobayashi, M. Function and characteristics of PINK1 in mitochondria. Oxid. Med. Cell Longev. 2013, 601587 (2013).
-
Zhang, L. et al. TRAP1 rescues PINK1 loss-of-function phenotypes. Hum. Mol. Genet. 22, 2829–2841 (2013).
-
Flinn, L. J. et al. TigarB causes mitochondrial dysfunction and neuronal loss in PINK1 deficiency. Ann. Neurol. 74, 837–847 (2013).
-
Gao, J. et al. Cytosolic PINK1 promotes the targeting of ubiquitinated proteins to the aggresome-autophagy pathway during proteasomal stress. Autophagy 12, 632–647 (2016).
-
Eldeeb, M. A. et al. Tom20 gates PINK1 activity and mediates its tethering of the TOM and TIM23 translocases upon mitochondrial stress. Proc. Natl Acad. Sci. USA 121, e2313540121 (2024).
-
Brown, S. J. et al. PINK1 deficiency impairs adult neurogenesis of dopaminergic neurons. Sci. Rep. 11, 6617 (2021).
-
Ryan, E., Seehra, G., Sharma, P. & Sidransky, E. GBA1-associated parkinsonism: new insights and therapeutic opportunities. Curr. Opin. Neurol. 32, 589–596 (2019).
-
Do, J., Perez, G., Berhe, B., Tayebi, N. & Sidransky, E. Behavioral phenotyping in a murine model of GBA1-associated Parkinson disease. Int. J. Mol. Sci. 22, 6826 (2021).
-
Bae, E.-J. et al. Loss of glucocerebrosidase 1 activity causes lysosomal dysfunction and α-synuclein aggregation. Exp. Mol. Med. 47, e153 (2015).
-
Abeliovich, A., Hefti, F. & Sevigny, J. Gene therapy for Parkinson’s disease associated with GBA1 mutations. J. Parkinsons Dis. 11, S183–S188 (2021).
-
Baden, P. et al. Glucocerebrosidase is imported into mitochondria and preserves complex I integrity and energy metabolism. Nat. Commun. 14, 1930 (2023).
-
Rosety, I. et al. Impaired neuron differentiation in GBA-associated Parkinson’s disease is linked to cell cycle defects in organoids. NPJ Parkinsons Dis. 9, 166 (2023).
-
Ungewickell, E. et al. Role of auxilin in uncoating clathrin-coated vesicles. Nature 378, 632–635 (1995).
-
Edvardson, S. et al. A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS ONE 7, e36458 (2012).
-
Köroğlu, Ç., Baysal, L., Cetinkaya, M., Karasoy, H. & Tolun, A. DNAJC6 is responsible for juvenile parkinsonism with phenotypic variability. Parkinsonism Relat. Disord. 19, 320–324 (2013).
-
Olgiati, S. et al. DNAJC6 mutations associated with early-onset Parkinson’s disease. Ann. Neurol. 79, 244–256 (2016).
-
Wulansari, N. et al. Neurodevelopmental defects and neurodegenerative phenotypes in human brain organoids carrying Parkinson’s disease-linked DNAJC6 mutations. Sci. Adv. 7, eabb1540 (2021).
-
Klein, C. & Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2, a008888 (2012).
-
Ahfeldt, T. et al. Pathogenic pathways in early-onset autosomal recessive Parkinson’s disease discovered using isogenic human dopaminergic neurons. Stem Cell Rep. 14, 75–90 (2020).
-
Morrone Parfitt, G. et al. Disruption of lysosomal proteolysis in astrocytes facilitates midbrain organoid proteostasis failure in an early-onset Parkinson’s disease model. Nat. Commun. 15, 447 (2024).
-
Wang, Q. et al. Molecular profiling of human substantia nigra identifies diverse neuron types associated with vulnerability in Parkinson’s disease. Sci. Adv. 10, eadi8287 (2024).
-
Monzel, A. S. et al. Machine learning-assisted neurotoxicity prediction in human midbrain organoids. Parkinsonism Relat. Disord. 75, 105–109 (2020).
-
Schober, A. Classic toxin-induced animal models of Parkinson’s disease: 6-OHDA and MPTP. Cell Tissue Res. 318, 215–224 (2004).
-
Meredith, G. E. & Rademacher, D. J. MPTP mouse models of Parkinson’s disease: an update. J. Parkinsons Dis. 1, 19–33 (2011).
-
Smeyne, R. J. & Jackson-Lewis, V. The MPTP model of Parkinson’s disease. Brain Res. Mol. Brain Res. 134, 57–66 (2005).
-
Schulz, J. B., Matthews, R. T., Muqit, M. M., Browne, S. E. & Beal, M. F. Inhibition of neuronal nitric oxide synthase by 7-nitroindazole protects against MPTP-induced neurotoxicity in mice. J. Neurochem. 64, 936–939 (1995).
-
Houlden, H. & Singleton, A. B. The genetics and neuropathology of Parkinson’s disease. Acta Neuropathol. 124, 325–338 (2012).
-
Pang, S. Y.-Y. et al. The interplay of aging, genetics and environmental factors in the pathogenesis of Parkinson’s disease. Transl. Neurodegener. 8, 23 (2019).
-
Logroscino, G. The role of early life environmental risk factors in Parkinson disease: what is the evidence? Environ. Health Perspect. 113, 1234–1238 (2005).
-
Ahmed, H., Abushouk, A. I., Gabr, M., Negida, A. & Abdel-Daim, M. M. Parkinson’s disease and pesticides: a meta-analysis of disease connection and genetic alterations. Biomed. Pharmacother. 90, 638–649 (2017).
-
Cannon, J. R. & Greenamyre, J. T. Gene-environment interactions in Parkinson’s disease: specific evidence in humans and mammalian models. Neurobiol. Dis. 57, 38–46 (2013).
-
Langston, J. W., Ballard, P., Tetrud, J. W. & Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219, 979–980 (1983).
-
Tetrud, J. W., Langston, J. W., Garbe, P. L. & Ruttenber, A. J. Mild parkinsonism in persons exposed to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Neurology 39, 1483–1487 (1989).
-
Brown, T. P., Rumsby, P. C., Capleton, A. C., Rushton, L. & Levy, L. S. Pesticides and Parkinson’s disease-is there a link? Environ. Health Perspect. 114, 156–164 (2006).
-
Rascol, O., Fabbri, M. & Poewe, W. Amantadine in the treatment of Parkinson’s disease and other movement disorders. Lancet Neurol. 20, 1048–1056 (2021).
-
Calabresi, P., Di Filippo, M., Ghiglieri, V., Tambasco, N. & Picconi, B. Levodopa-induced dyskinesias in patients with Parkinson’s disease: filling the bench-to-bedside gap. Lancet Neurol. 9, 1106–1117 (2010).
-
Stephen, P. J. & Williamson, J. Drug-induced parkinsonism in the elderly. Lancet 2, 1082–1083 (1984).
-
Bhaduri, A. et al. Cell stress in cortical organoids impairs molecular subtype specification. Nature 578, 142–148 (2020).
-
Xiang, Y. et al. hESC-derived thalamic organoids form reciprocal projections when fused with cortical organoids. Cell Stem Cell 24, 487–497.e7 (2019).
-
Zafeiriou, M.-P. et al. Developmental GABA polarity switch and neuronal plasticity in Bioengineered Neuronal Organoids. Nat. Commun. 11, 3791 (2020).
-
Jenner, P. & Olanow, C. W. Oxidative stress and the pathogenesis of Parkinson’s disease. Neurology 47, S161–S170 (1996).
-
Pamies, D. et al. Human IPSC 3D brain model as a tool to study chemical-induced dopaminergic neuronal toxicity. Neurobiol. Dis. 169, 105719 (2022).
-
Renner, H. et al. Cell-type-specific high throughput toxicity testing in human midbrain organoids. Front. Mol. Neurosci. 14, 715054 (2021).
-
Dorgau, B. et al. Human retinal organoids provide a suitable tool for toxicological investigations: a comprehensive validation using drugs and compounds affecting the retina. Stem Cells Transl. Med. 11, 159–177 (2022).
-
Katzenschlager, R. & Lees, A. J. Treatment of Parkinson’s disease: levodopa as the first choice. J. Neurol. 249(Suppl 2), II19–II24 (2002).
-
Bronstein, J. M. et al. Deep brain stimulation for Parkinson disease: an expert consensus and review of key issues. Arch. Neurol. 68, 165 (2011).
-
Faggiani, E. & Benazzouz, A. Deep brain stimulation of the subthalamic nucleus in Parkinson’s disease: from history to the interaction with the monoaminergic systems. Prog. Neurobiol. 151, 139–156 (2017).
-
Kim, M. S., Kim, H. & Lee, G. Precision medicine in Parkinson’s disease using induced pluripotent stem cells. Adv. Healthc. Mater. e2303041 https://doi.org/10.1002/adhm.202303041 (2024).
-
Adler, A. F. et al. hESC-derived dopaminergic transplants integrate into basal ganglia circuitry in a preclinical model of Parkinson’s disease. Cell Rep. 28, 3462–3473.e5 (2019).
-
Kikuchi, T. et al. Human iPS cell-derived dopaminergic neurons function in a primate Parkinson’s disease model. Nature 548, 592–596 (2017).
-
Mansour, A. A. et al. An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 36, 432–441 (2018).
-
Fiorenzano, A. et al. Single-cell transcriptomics captures features of human midbrain development and dopamine neuron diversity in brain organoids. Nat. Commun. 12, 7302 (2021).
-
Birtele, M. et al. Single-cell transcriptional and functional analysis of dopaminergic neurons in organoid-like cultures derived from human fetal midbrain. Development 149, dev200504 (2022).
-
Devine, M. J. et al. Parkinson’s disease induced pluripotent stem cells with triplication of the α-synuclein locus. Nat. Commun. 2, 440 (2011).
-
Zheng, X. et al. Human iPSC-derived midbrain organoids functionally integrate into striatum circuits and restore motor function in a mouse model of Parkinson’s disease. Theranostics 13, 2673–2692 (2023).
-
Jarazo, J. et al. Parkinson’s disease phenotypes in patient neuronal cultures and brain organoids improved by 2-hydroxypropyl-β-cyclodextrin treatment. Mov. Disord. 37, 80–94 (2022).
-
Kim, M. S. et al. Advanced human iPSC-based preclinical model for Parkinson’s disease with optogenetic α-synuclein aggregation. Cell Stem Cell 30, 973–986.e11 (2023).
-
Shin, M. et al. Human motor system-based biohybrid robot-on-a-chip for drug evaluation of neurodegenerative disease. Adv. Sci. 11, e2305371 (2024).
-
Swinney, D. C. & Anthony, J. How were new medicines discovered? Nat. Rev. Drug Discov. 10, 507–519 (2011).
-
Eder, J., Sedrani, R. & Wiesmann, C. The discovery of first-in-class drugs: origins and evolution. Nat. Rev. Drug Discov. 13, 577–587 (2014).
-
Gribkoff, V. K. & Kaczmarek, L. K. The need for new approaches in CNS drug discovery: why drugs have failed, and what can be done to improve outcomes. Neuropharmacology 120, 11–19 (2017).
-
Swalley, S. E. Expanding therapeutic opportunities for neurodegenerative diseases: a perspective on the important role of phenotypic screening. Bioorg. Med. Chem. 28, 115239 (2020).
-
Friese, A. et al. The convergence of stem cell technologies and phenotypic drug discovery. Cell Chem. Biol. 26, 1050–1066 (2019).
-
Boussaad, I. et al. Integrated, automated maintenance, expansion and differentiation of 2D and 3D patient-derived cellular models for high throughput drug screening. Sci. Rep. 11, 1439 (2021).
-
Rimann, M. & Graf-Hausner, U. Synthetic 3D multicellular systems for drug development. Curr. Opin. Biotechnol. 23, 803–809 (2012).
-
Edmondson, R., Broglie, J. J., Adcock, A. F. & Yang, L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay. Drug Dev. Technol. 12, 207–218 (2014).
-
Montanez-Sauri, S. I., Beebe, D. J. & Sung, K. E. Microscale screening systems for 3D cellular microenvironments: platforms, advances, and challenges. Cell Mol. Life Sci. 72, 237–249 (2015).
-
Grenier, K., Kao, J. & Diamandis, P. Three-dimensional modeling of human neurodegeneration: brain organoids coming of age. Mol. Psychiatry 25, 254–274 (2020).
-
Babu, H. W. S., Kumar, S. M., Kaur, H., Iyer, M. & Vellingiri, B. Midbrain organoids for Parkinson’s disease (PD)—a powerful tool to understand the disease pathogenesis. Life Sci. 345, 122610 (2024).
-
Kortekaas, R. et al. Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann. Neurol. 57, 176–179 (2005).
-
de Rus Jacquet, A. et al. The contribution of inflammatory astrocytes to BBB impairments in a brain-chip model of Parkinson’s disease. Nat. Commun. 14, 3651 (2023).
-
Han, L. & Jiang, C. Evolution of blood-brain barrier in brain diseases and related systemic nanoscale brain-targeting drug delivery strategies. Acta Pharm. Sin. B 11, 2306–2325 (2021).
-
Cakir, B. et al. Engineering of human brain organoids with a functional vascular-like system. Nat. Methods 16, 1169–1175 (2019).
-
Daviaud, N., Friedel, R. H. & Zou, H. Vascularization and engraftment of transplanted human cerebral organoids in mouse cortex. eNeuro 5, ENEURO.0219-18.2018 (2018).
-
Dao, L. et al. Modeling blood-brain barrier formation and cerebral cavernous malformations in human PSC-derived organoids. Cell Stem Cell 31, 818–833.e11 (2024).
-
Czerniecki, S. M. et al. High-throughput screening enhances kidney organoid differentiation from human pluripotent stem cells and enables automated multidimensional phenotyping. Cell Stem Cell 22, 929–940.e4 (2018).
-
Ao, Z. et al. One-stop microfluidic assembly of human brain organoids to model prenatal cannabis exposure. Anal. Chem. 92, 4630–4638 (2020).
-
Nayak, D., Roth, T. L. & McGavern, D. B. Microglia development and function. Annu. Rev. Immunol. 32, 367–402 (2014).
-
Takasato, M. et al. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature 526, 564–568 (2015).
-
Sabate-Soler, S. et al. Microglia integration into human midbrain organoids leads to increased neuronal maturation and functionality. Glia 70, 1267–1288 (2022).
-
Zhang, C., Bakker, A. D., Klein-Nulend, J. & Bravenboer, N. Studies on osteocytes in their 3D native matrix versus 2D in vitro models. Curr. Osteoporos. Rep. 17, 207–216 (2019).
-
Pais, A. S. et al. The challenge of ovarian tissue culture: 2D versus 3D culture. J. Ovarian Res. 14, 147 (2021).
-
Marín, O., Yaron, A., Bagri, A., Tessier-Lavigne, M. & Rubenstein, J. L. Sorting of striatal and cortical interneurons regulated by semaphorin-neuropilin interactions. Science 293, 872–875 (2001).
-
Kwan, K. Y., Sestan, N. & Anton, E. S. Transcriptional co-regulation of neuronal migration and laminar identity in the neocortex. Development 139, 1535–1546 (2012).
-
Clowry, G. J. et al. Charting the protomap of the human telencephalon. Semin Cell Dev. Biol. 76, 3–14 (2018).
-
Molnár, Z. et al. New insights into the development of the human cerebral cortex. J. Anat. 235, 432–451 (2019).
-
López-Bendito, G. & Molnár, Z. Thalamocortical development: how are we going to get there? Nat. Rev. Neurosci. 4, 276–289 (2003).
-
Renner, M. et al. Self-organized developmental patterning and differentiation in cerebral organoids. EMBO J. 36, 1316–1329 (2017).
-
Eiraku, M. et al. Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem Cell 3, 519–532 (2008).
-
Muguruma, K. et al. Ontogeny-recapitulating generation and tissue integration of ES cell-derived Purkinje cells. Nat. Neurosci. 13, 1171–1180 (2010).
-
Nakano, T. et al. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 10, 771–785 (2012).
-
Kadoshima, T. et al. Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc. Natl Acad. Sci. USA 110, 20284–20289 (2013).
-
Marton, R. M. & Pașca, S. P. Organoid and assembloid technologies for investigating cellular crosstalk in human brain development and disease. Trends Cell Biol. 30, 133–143 (2020).
-
Qian, X. et al. Generation of human brain region-specific organoids using a miniaturized spinning bioreactor. Nat. Protoc. 13, 565–580 (2018).
-
Dong, X. et al. Human cerebral organoids establish subcortical projections in the mouse brain after transplantation. Mol. Psychiatry 26, 2964–2976 (2021).
-
Moriarty, N. et al. A combined cell and gene therapy approach for homotopic reconstruction of midbrain dopamine pathways using human pluripotent stem cells. Cell Stem Cell 29, 434–448.e5 (2022).
-
Shrigley, S. et al. Grafts derived from an α-synuclein triplication patient mediate functional recovery but develop disease-associated pathology in the 6-OHDA model of Parkinson’s disease. J. Parkinsons Dis. 11, 515–528 (2021).
-
Li, C. et al. Single-cell brain organoid screening identifies developmental defects in autism. Nature 621, 373–380 (2023).
-
Chiaradia, I. et al. Tissue morphology influences the temporal program of human brain organoid development. Cell Stem Cell 30, 1351–1367.e10 (2023).
-
Yan, Y. et al. 3D bioprinting of human neural tissues with functional connectivity. Cell Stem Cell 31, 260–274.e7 (2024).
-
Geng, L. et al. MLKL deficiency alleviates neuroinflammation and motor deficits in the α-synuclein transgenic mouse model of Parkinson’s disease. Mol. Neurodegener. 18, 94 (2023).
-
Hendriks, D. et al. Human fetal brain self-organizes into long-term expanding organoids. Cell 187, 712–732.e38 (2024).
Acknowledgements
The authors’ work was supported by the National Natural Science Foundation of China (82271277 to C.M.), and the Innovative and Scientific and Technological Talent Training Project of Henan Province (Grant YXKC2021062 to C.M.), and the Non-profit Central Research Institute Fund of Chinses Academy of Medical Sciences (2020-PT310-01 to Y.X.).
Author information
Authors and Affiliations
Contributions
Xin Cui, Xinwei Li, Huimin Zheng and Yun Su drafted and revised of the manuscript for content, including medical writing for the content. Changhe Shi, Shuyu Zhang, Mengjie Li, Xiaoyan Hao, Shuo Zhang, Zhengwei Hu, Zongping Xia and Changhe Shi assembled and edited the figures and tables. Chengyuan Mao and Yuming Xu conceived the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cui, X., Li, X., Zheng, H. et al. Human midbrain organoids: a powerful tool for advanced Parkinson’s disease modeling and therapy exploration. npj Parkinsons Dis. 10, 189 (2024). https://doi.org/10.1038/s41531-024-00799-8
-
Received:
-
Accepted:
-
Published:
-
DOI: https://doi.org/10.1038/s41531-024-00799-8
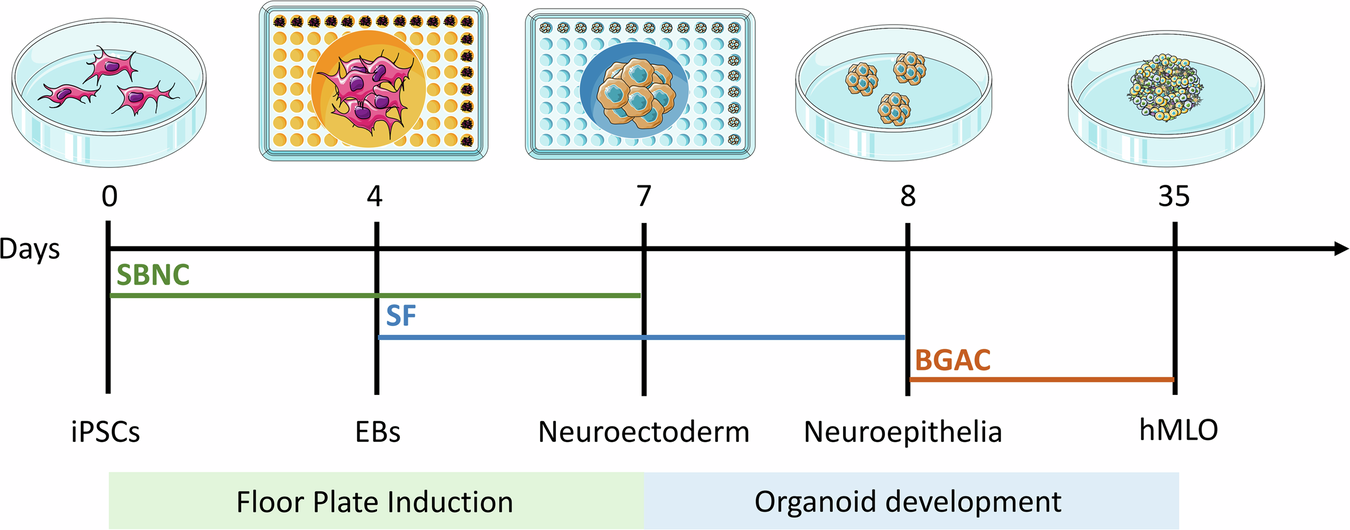
This news item came from: https://www.nature.com/articles/s41531-024-00799-8

